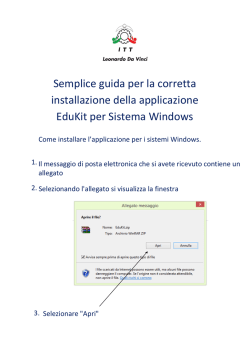
1 GESTIONE DEL RISCHIO NEI DISPOSITIVI MEDICI: DALLA CLASSIFICAZIONE ALLA COMMERCIALIZZAZIONE Ing. Enrico Perfler – Eudax s.r.l. Milano, 23 Gennaio 2014 Indice 2 Il concetto di rischio nei dispositivi medici ¨ La normativa vigente ¨ I Requisiti Essenziali ¨ I “limiti” della Direttiva ¨ Lo standard ISO 14971per la gestione del rischio ¨ Conclusioni ¨ Il rischio nei dispositivi medici 3 Il concetto di rischio nei dispositivi medici è collegato ad alcuni concetti fondamentali: ¨ Destinazione d’uso ¨ Invasività della procedura ¨ Durata trattamento/ tempo d’applicazione del dispositivo medico ¨ Principio d’azione ¨ Materiali, tecnologia, ecc. … Il rischio nei dispositivi medici 4 Classificazione del dispositivo medico secondo allegato IX della Direttiva 93/42/CEE e ss.mm.ii. Il rischio nei dispositivi medici 5 Processo produttivo ¨ Uso combinato con altri dispositivi medici, farmaci, ecc. ¨ Ambiente di utilizzo del prodotto ¨ Utente finale ¨ … ¨ La normativa vigente 6 Classificazione (Considerando N.15, Allegato IX) considerando che, in particolare in vista delle procedure di valutazione della conformità, è necessario suddividere i dispositivi medici in quattro classi di prodotti; che le regole di decisione di classificazione si basano sulla vulnerabilità del corpo umano e tengono conto dei rischi potenziali connessi con l'elaborazione tecnologica dei dispositivi e con la loro fabbricazione… Requisiti Essenziali (Articolo 3, Allegato I) I dispositivi devono soddisfare i pertinenti requisiti essenziali prescritti nell'allegato I in considerazione della loro destinazione. La normativa vigente 7 Valutazione clinica (Art.15, Allegato X) La conferma del rispetto dei requisiti relativi alle caratteristiche e alle prestazioni specificate ai punti 1 e 3 dell’allegato I in condizioni normali di utilizzazione del dispositivo, nonché la valutazione degli effetti collaterali e dell’accettabilità del rapporto rischi/benefici di cui al punto 6 dell’allegato I devono basarsi, in linea di principio, su dati clinici. (…) Sorveglianza Allegato II – Dichiarazione CE di Conformità Allegato V – Garanzia di qualità della produzione Allegato VI – Garanzia di qualità del prodotto Allegato X – Valutazione clinica Requisiti Essenziali 8 ¨ ¨ I requisiti essenziali sono descritti nell’Allegato I della Direttiva 93/42/CEE. Essi rappresentano le prescrizioni di minima in termini di caratteristiche intrinseche di ogni prodotto (chimiche, fisiche, biologiche, ecc.), di specifiche costruttive, di prestazioni funzionali, di presentazione (confezione, etichettatura), ecc. cui i dispositivi devono essere assoggettati nella progettazione e nella fabbricazione per poter essere definiti conformi, in considerazione della loro destinazione d'uso. Requisiti Essenziali 9 ¨ I Requisiti Essenziali previsti dalla Direttiva rappresentano gli elementi il cui rispetto giustifica la ragionevole aspettativa che il prodotto svolga le funzioni per le quali è stato progettato, nel modo descritto dal fabbricante. Allegato I Direttiva 10 ¨ ¨ REQUISITI GENERALI REQUISITI RELATIVI ALLA PROGETTAZIONE E ALLA COSTRUZIONE ¤ Caratteristiche chimiche, fisiche e biologiche ¤ Infezione e contaminazione microbica ¤ Caratteristiche relative alla fabbricazione e all’ambiente ¤ Dispositivi con funzioni di misura ¤ Protezione contro le radiazioni ¤ Requisiti per i dispositivi medici collegati o dotati di una fonte di energia ¤ Informazioni fornite dal fabbricante “Limiti” della Direttiva 11 ¨ La Direttiva Europea per i dispositivi medici riporta “unicamente” i Requisiti Essenziali volti a garantire che i dispositivi siano sicuri e che la loro funzionalità sia in accordo a quanto specificato dal fabbricante. non descrive i requisiti tecnici o le modalità di test applicabili in generale o a singole tipologie di dispositivi Rinvio alle norme 12 Rinvio alle norme (Direttiva, Art. 5) Nel sistema normativo Europeo, se il fabbricante è in grado di dimostrare di aver adeguatamente applicato le norme armonizzate rilevanti (anche denominate standard), questo implica una presunzione di conformità ai requisiti della Direttiva. A fronte del carattere generale e non tecnico di ciascuno dei requisiti della Direttiva, le norme armonizzate contengono specifiche tecniche dettagliate sugli elementi da implementare per essere in conformità con le medesime. Norme armonizzate 13 ¨ ¨ Gli standard armonizzati o norme armonizzate contengono specifiche tecniche applicabili in una vasta gamma di aree di interesse e rappresentano uno strumento applicabile per dimostrare la conformità ai requisiti essenziali della Direttiva. Essi spesso derivano da Standard Internazionali, che ne rappresentano il progenitore e che vengono recepiti in parte od integralmente. UNI EN ISO 14971:2012 14 Applicazione della gestione dei rischi ai dispositivi medici ¨ La norma specifica una procedura che permette al fabbricante di identificare i pericoli associati ai dispositivi medici, inclusi i dispositivi medico-diagnostici in vitro, per stimare e valutare i rischi associati, per controllare tali rischi, e per monitorare l'efficacia dei controlli. I requisiti della norma sono applicabili a tutte le fasi del ciclo di vita di un dispositivo medico. La norma non si applica al processo decisionale clinico e non specifica i livelli di rischio accettabili. Il rischio nei dispositivi medici Gravità 15 Utilizzo Installazione Produzione Design Tempo Struttura della norma ISO 14971 16 ¨ ¨ ¨ ¨ ¨ ¨ ¨ ¨ ¨ Sezione 1 – Scopo Sezione 2 – Termini e definizioni Sezione 3 – Requisiti Generali per la Gestione del Rischio Sezione 4 – Analisi del Rischio Sezione 5 – Valutazione del Rischio Sezione 6 – Controllo del Rischio Sezione 7 – Valutazione dell’accettabilità del rischio residuo globale Sezione 8 – Report della Gestione del Rischio Sezione 9 – Informazioni di Produzione e Post-Produzione Processo di gestione dei rischi 17 ¨ Il Fabbricante deve stabilire, documentare e mantenere per tutto il ciclo di vita, un processo per identificare i pericoli associati ad un dispositivo medico, stimando e valutando i rischi associati, controllando gli stessi e monitorando l’efficienza dei controlli. 18 Responsabilità 19 3.2 Responsabilità della Direzione La Direzione deve fornire prova del suo impegno nel processo di gestione del rischio: ¨ ¤ Assicurando la disponibilità di adeguate risorse ¤ Assicurando l’assegnazione di personale qualificato per la gestione del rischio ¤ Definendo e documentando una politica per la determinazione dei criteri riferiti all’accettabilità del rischio ¤ Revisionando ad intervalli prestabiliti l’idoneità del processo di gestione dei rischi Piano di gestione del rischio 20 ¨ ¨ ¨ ¨ ¨ ¨ L’obiettivo delle attività pianificate di gestione del rischio, identificando e descrivendo il dispositivo medico e il ciclo di vita per i quali ogni elemento del Piano è applicabile Assegnazione di ruoli e attività Requisiti per la revisione delle attività di gestione del rischio Criteri per l'accettabilità del rischio, basati sulla politica del Fabbricante per la determinazione dei rischi Attività di verifica Attività relative alla raccolta e revisione delle informazioni rilevanti di produzione e post-produzione (Post-Market Clinical Follow-Up) Risk management file 21 Per il particolare dispositivo medico o accessorio preso in considerazione, i risultati di tutte le attività di gestione del rischio devono essere registrate e mantenute nella documentazione di gestione del rischio. Per ogni pericolo identificato, la documentazione di gestione del rischio deve fornire la tracciabilità rispetto a: ¨ Analisi del rischio ¨ Valutazione del rischio ¨ Implementazione e verifica delle misure di controllo ¨ Accettabilità del rischio residuo Processo analisi dei rischi 22 Stima del rischio 23 ¨ La stima del rischio è un processo utilizzato per quantificare la probabilità di occorrenza di danni e la gravità di questi. Indice di rischio 24 Il concetto di rischio è la combinazione delle due componenti seguenti: ¨ la probabilità del verificarsi del danno, ovvero, con che frequenza può verificarsi il danno; ¨ le conseguenze di tale danno, ovvero, la gravità dello stesso. Probabilità X Severità = RI (Risk Index) Probabilità X Severità X Detection = RPN (Risk Prority Number) Stima della probabilità 25 Stima della gravità 26 Accettazione del rischio 27 Accettazione del rischio 28 Accettazione del rischio 29 Controllo del rischio 30 ¨ L’intervento di controllo del rischio può avvenire: ¤ nella revisione del progetto (sicurezza intrinseca del DM) ¤ nella produzione del dispositivo ¤ nel fornire formazione agli utenti ¤ nel fornire informazioni sull’uso Swine flu: don’t do this! 31 Controllo dei rischi residui 32 ¨ Gli effetti delle misure di controllo del rischio devono essere revisionati in merito a: ¤ introduzione di nuovi potenziali pericoli o situazioni di pericolo; ¤ influenza sulla stima dei rischi precedentemente analizzati (migliorativa o peggiorativa) ¨ Ogni nuovo rischio, ed ogni rischio modificato dalle misure di controllo, dovrà essere gestito come descritto nel piano di gestione dei rischi. Accettazione del rischio 33 Valutazione rischio vs. benefici 34 ¨ ¨ ¨ Se il rischio è giudicato non accettabile utilizzando i criteri del piano di gestione dei rischi ed ulteriori controlli non sono applicabili, il fabbricante può raccogliere e revisionare i dati e la letteratura scientifica per determinare se i benefici clinici superano il rischio residuo. Se l’evidenza clinica non supporta la conclusione che i benefici clinici superano il rischio residuo, allora il rischio rimane inaccettabile. Diversamente, per i rischi che risultano essere inferiori ai benefici, il Fabbricante decide quali informazioni per la sicurezza sono necessari al fine di mitigare il rischio residuo. Informazioni di produzione e post-produzione 35 ¨ ¨ Il fabbricante deve stabilire, documentare e mantenere un sistema per raccogliere e revisionare le informazioni sul DM o su simili DM durante la fase produttiva e postproduttiva (commercializzazione, riparazione, ecc.). Nel mettere in atto un sistema di revisione di queste informazioni, il fabbricante dovrà inoltre considerare: ¤ il metodo di raccolta e revisione delle informazioni generate dall’operatore, l’utente, durante l’installazione, l’uso e la manutenzione del DM; ¤ gli standard disponibili (nuovi o revisionati) 36 Grazie per la cortese attenzione!
© Copyright 2025 Paperzz