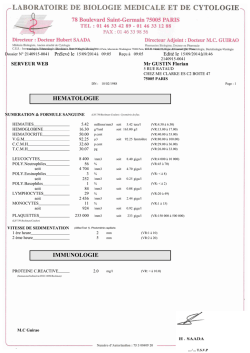
Bartter et Gitelman jouent aux diurétiques Rosa Vargas-Poussou Département de Génétique Hôpital Européen Georges Pompidou Paris Syndromes de Bartter et Gitelman Tubulopathies héréditaires rares Transmission autosomique récessive Prévalence Syndrome de Gitelman: 1/40.000 Syndrome de Bartter: 1/1.000.000 Défaut de réabsorption de sodium dans le néphron distal Syndromes de Bartter et Gitelman Caractéristiques cliniques communes Activation secondaire du système rénine angiotensine - aldostérone Alcalose métabolique Hypokaliémie d’origine rénale Pression artérielle normale ou basse Tubule Rénal - Réabsorption Tube Proximal Na+ 60% Ca+2 60% Branche large ascendante de l’anse d’Henle Mg+2 70% Na+ 20-25% Ca+2 25-30% Tube contourné distal Na+ 7% Ca+2 et Mg+2 10% Tube collecteur Na+ 2% H2O Sécrétion H+ et K + Tube proximal Tube collecteur Acétazolamide H+ HCO3- Na+ H+ ACIV ACII H+ H2O CO2 Glucose Phosphate Acides aminés Na+ 3HCO3- HCO3- Pendrine Na+ Glutamine Na+ NH4+ NH4+ NH3 H+ NH3 Na+ K+ HCO3- Glutamate Cl- cetoglutarate Glucose NH4+ HCO3- H2O CO2 Furosémide K+ H+ Ca++ Mg++ NH4+ (+) K+ ClCl- H+ HCO3- Intercalaire B Na + HCO3- NDCBE K+ Na+ K+ Cl - H+ Cl K+ Na+ Na+ HCO3Cl- Acétazolamide Barttine HCO3 H+ H+ Branche large – Anse d’Henle 2Cl Na+ K+ Na + Cl- Intercalaire A ACII K+ Na + K+ CO2 HCO3- H2O (-) (+) ENaC Amiloride Claudines Na + Na + K+ (-) Principale Vte Aldostérone K+ Cl- Cl Na+ Thiazides Na+ K+ Ca2+ Mg2+ Na+ H+ Na+ Na+ Na+ Ca2+ H2O RM Ca2+ Mg2+ Tube contourné distal Spironolactone Eplérenone H2O Syndromes de Bartter et Gitelman Na+ 7% Cl Na+ Thiazides Na+ Na+ Mg2+ K+ Na+ Ca2+ Mg2+ H+ ClNa+ Ca2+ Ca2+ Na+ Na+ 20% 2Cl Na+ K+ Furosemide K+ K+ Ca++ Mg++ NH4+ (+) H+ Na+ Na+ Vte Na+ K+ Cl Barttin Cl K+ Adolescent Adulte Hypomagnésémie Hypocalciurie Nourrisson - enfant Normo ou hypercalciurie Polyurie Retard de croissance Gitelman Bartter classique Hydramnios Polyurie sévère Bartter Antenatal Retard de (-) croissance Hypercalciurie Néphrocalcinose PG Parfois surdité HCO3- Syndromes de Bartter et Gitelman Na+ 7% Cl Na+ Thiazides ClK+ Na+ Na+ Ca2+ Ca2+ Mg2+ Na+ H+ Na+ 20% Bartter type I (anténatal) 2Cl Na+ K+ Furosémide Bartter type II (anténatal) Na+ Ca2+ Na+ Mg2+ K+ K+ Ca++ Mg++ NH4+ (+) H+ Na+ Gitelman Na+ K+ Cl Barttin Cl K+ Bartter type IV (anténatal+surdité) HCO3- Na+ Bartter (-) Vte Bartter type III (classique) Na+ 7% 2Cl Na+ K+ Furosémide K+ HCO3- Cl K+ Na+ Na+ Ca++ Mg++ NH4+ HCO3- Barttine K+ H+ Pendrine Na+ K+ Cl - Cl- HCO3- K+ ClCl- H+ HCO3- Na+ 2% (-) NDCBE HCO3 H+ H+ Vte HCO3Cl- Intercalaire A ACII Hypercalciurie Polyurie H+ K+ Na + K+ CO2 H2O (-) Na+ K+ Ca2+ Mg2+ Na+ H+ Na+ Na+ Na+ (+) ENaC Cl- Cl Na+ Thiazides Intercalaire B Na + Na+ 20% Claudines (+) Na + Cl- Amiloride Na + Na + K+ Ca2+ Ca2+ Mg2+ Alcalose Hypokaliémie Principale Aldostérone K+ H2O RM Spironolactone Eplérenone H2O Na+ 7% 2Cl Na+ K+ Furosémide K+ HCO3- Cl K+ Na+ Na+ Ca++ Mg++ NH4+ HCO3- Barttine K+ H+ Pendrine Na+ K+ Cl - Cl- HCO3- K+ ClCl- H+ HCO3- Na+ 2% (-) NDCBE HCO3 H+ H+ Vte HCO3Cl- Intercalaire A ACII Normo ou hypercalciurie Polyurie H+ K+ Na + K+ CO2 H2O (-) Na+ K+ Ca2+ Mg2+ Na+ H+ Na+ Na+ Na+ (+) ENaC Cl- Cl Na+ Thiazides Intercalaire B Na + Na+ 20% Claudines (+) Na + Cl- Amiloride Na + Na + K+ Ca2+ Ca2+ Mg2+ Alcalose Hypokaliémie Principale Aldostérone K+ H2O RM Spironolactone Eplérenone H2O Na+ 7% 2Cl Na+ K+ Furosémide K+ HCO3- Cl K+ Na+ Na+ Ca++ Mg++ NH4+ HCO3- Barttine K+ H+ Pendrine Na+ K+ Cl - Cl- HCO3- K+ ClCl- H+ HCO3- Intercalaire B Na + Na+ 20% Claudines Na+ 2% (-) (+) Na + Cl- NDCBE HCO3 H+ H+ Vte HCO3Cl- Intercalaire A ACII H+ K+ Na + K+ CO2 H2O (-) Cl Na+ Thiazides Na+ K+ Ca2+ Mg2+ Na+ H+ Na+ Na+ Na+ (+) ENaC Cl- Amiloride Na + Na + K+ Ca2+ Ca2+ Mg2+ Alcalose Hypokaliémie Principale Aldostérone K+ H2O RM Hypomagnésémi e Hypocalciurie Spironolactone Eplérenone H2O Syndrome de Gitelman - Population Janvier 2001 à août 2009 448 patients avec diagnostic clinique de SG (219 H et 229 F) Réseau Français de Tubulopathies et Eunefron Critères – – – – – – Hypokaliémie d’origine rénale Alcalose métabolique Fuite sodée Activation secondaire du système RAA Hypomagnésémie Hypocalciurie SLC12A3 – Séquençage MUTATIONS 172 différentes (100 nouvelles) 6% 2% 14% 15% 70% (n=315) 63% Missense (n=110) Frameshift (n=26) Splicing (n=24) Nonsense (n=11) Inframe (n=4) 52 patients sans mutations Analyse du gène CLCNKB chez 49 patients: détection d’anomalies moléculaires chez 14 38 patients sans anomalies moléculaires (8.4%) MLPA (Multiplex Ligation-dependant Probe QMPSF Amplification) Quantitative Multiplex PCR Short Fluorescent Fragments 6-FAM Forward primer 5’ Exon 3’ « queue » M13 Reverse primer Salsa® MLPA® kit P136 SLC12A3 Gitelman Syndrome MRC Holland 38 sondes: 25/26 exons du gène SLC12A3 (excepte exon 6) and 13 contrôl Rapport < 0.7 délétion 0.7 - 1.3 Normal 1.3 – 2 duplication Résultats MLPA 11 grands réarrangements hétérozygotes chez 24/51 patients du groupe avec une seule mutation hétérozygote – Délétion d’un exon chez 13 patients: • • • • E9del, 1 patient E14del, 2 patients E18del, 1 patient E26del, 9 patients – Délétions ou duplications de 2 ou plus exons chez 11 patients • • • • • • • E1_E7del, 2 patients E2_E3del, 2 patients E4_E6del, E4_E5del, 3 patients E19_E23del, 1 patient E24_E25del, 1 patient E1_E3dup, 1 patient E1_E4dup, 1 patient 1 2 3 4 5 6 7 8 9 10 11 12 13 (3342 bp) 14 19 20 21 22 23 24 25 AluSx 26 (1183 bp) L2b_LINE AluJb L2a_LINE 15 16 17 18 (2720 bp) MER115 AluY AluSx AluSx AluSx AluSx Grands réarrangements – Gène SLC12A3 (10711bp) (1355 bp) (2416bp) (1508 bp) AluSx AluSx AluY MER115 Intron 1 5’ CTACTTGCTTATCACCGTGGCTCTGTGAGGACTGGGGACACAATCTG patient CTACTTGCTTATCACCGTGGCTCTGAAGGCAGTAAAGTGGGGTGATG GCTGTTGGGACTGTGGAGGGCTCTGAAGGCAGTAAAGTGGGGTGATG 3’ Intron 13 Intron 3 Intron 14 5’ TAAAGAGGCTTGAGGAAGACTTTTTCTTTCTTTTTTTTTTTT patient TAAAGAGGCTTGAGGAAGACCCTGAGTGAGCTTCCAGGGCCT 3’ GAATCCCCTGTCCGAAGGACCCTGAGTGAGCTTCCAGGGCCT 5’ GGGTAGGGCTTGTCCCAGGTGAAGCTTTGTGGATGGAACTTCCAAGTGTGACATAGCTGTTTAGTATCCCAGTTACCCTTCTCAGAGGAG patient GGGTAGGGCTTGTCCCAGGTGAAGCTTTTTAGTAGAGATGGGGTTTAGTAGAGATTTTTAGTAGAGATGGGGTTTCACCATGTTGACCAG 3’ CTCCCGAATAGCTGGGATTACAGGCACCTGCCATCACACGAGCTAATTTTTGTATTTTTAGTAGAGATGGGGTTTCACCATGTTGACCAG Deletions 1-3 et 5: points de cassure dans Alu(s) avec homologie > 80% Non-allelic homologous recombination (NAHR) Deletions 4 et 6: points de cassure dans LCRs non homologues Non-homologous end-joining (NHEJ) Deletion 7: E26del: point de cassure en 5’ proche d’un AluJb et point de cassure en 3’ dans un AluSx Mécanisme complexe Séquençage + Recherche de grands réarrangements 6% 2% 6% 13% 14% Taux de détection de mutations de 91% 59% Missense (n=110) Frameshift (n=26) Splicing (n=24) Nonsens (n=11) Inframe (n=4) Grands réarrangements(n=11) Vargas-Poussou R, JASN 2011 SLC12A3 - Mutations ponctuelles Fréquence et distribution - 172 mutations - 711 allèles Kunchaparty, S, et al. Am J Physiol, 1999. De Jong, JC, et al. J Am Soc Nephrol, 2002. Cotransporteur Na+-Cl * * ** *Thr46, Thr55, and Thr60 4 3 TGN Golgi ERGIC 2 RE Ubiquitineprotéasome 1 Séquençage + Recherche de grands réarrangements Janvier 2001 à août 2012 662 cas index 12% 6%2% 7% 3% 13% 14% 55% 14% 58% 16% Hétérozygotes composites (n = 360) Homozygotes (n = 105) Une mutation hétérozygote (n = 94) Pas de mutation (n = 81) CLCNKB (n=22) Taux de détection de mutations de 88% Missense (n=130) Frameshift (n=31) Splicing (n=28) Nonsens (n=13) Inframe (n=4) Grands réarrangements(n=16) Base de données - Syndrome Gitelman Janvier 2012 • 375 patients : 189 M et 186 F • 3 Centres 9.5% – Paris : 314 – Brussels: 32 patients – Nijmegen: 29 patients • Mutations – SLC12A3: 363 – CLCNKB: 12 16% 7% n= 348 Med IQR: 25 (14-38) Symptoms at diagnosis 25 20 15 % 10 5 0 Fortuitous cramps Asthenia Failure to Thrive Polyuria Tetany Arrhytmia Malaise Chondrocalcinosis Vomiting Paresia and paresthesia Hypotonia Abdominal pain Enuresis constipation Donnés biologiques au diagnostic n= 250 Med IQR: 138.8 (137-140) n= 280 Med IQR 2.37 (2.25-2.47) n= 41 Med IQR 286 (240-329) Donnés biologiques au diagnostic 39% 22 % n= 322 Med IQR: 0.62 (0.52-0.68) n= 151 Med IQR 0.08 (0.03-0.18) Syndrome de Bartter de type III Na+ 2Cl K+ K+ Na+ K+ Cl - Na+ K+ Ca2+ Mg2+ Na+ Na+ H+ Cl - Na+ (+) Mg++ NH4+ Ca2+ Ca2+ Mg2+ Tube contourné distal RSCa Na+ Ca++ Na+ Na+ K+ K+ H+ Cl- Cl Na+ Thiazides HCO3- Pendrine HCO3HCO3- Claudins 16 - 19 (-) Cl- Cl- Na + K+ ClCl- HCO3Na + NDCBE TC – C. Intercalaires B SB Anténatal SB Classique S de Gitelman Alcalose Hypochlorémique ++++ H+ CLCNKA-CLCNKB – 1p36 Bartter type 3 Missense 12% N=91 8% Nonsense Homozygous Compound htz 1 htz mutation n=43 n=34 n=14 Splicing 10% 55% 15% Frameshift Large rearrangements N Homozygous whole gene deletion Antenatal Bartter 23 11 (45%) Classic Bartter 47 8 (17%) Gitelman 24 2 (8.3%) Expression fonctionnelle de mutations du gène CLCNKB Keck M et al. Human Mutation, 2013 Andrini O et al. Pflugers Arch. 2013 n=94 17.5 ans IQR(8-36) n=35 1an IQR(0.4-3) p<0.0001 SLC12A3 Pédiatrie SLC12A3 Adultes CLCNKB Fortuite 58% 40% 3% Retard de croissance 18% 5% 60% Faiblesse musculaire 9% 5% 9.6% Asthénie 9% 32.5% Tétanie 9% 7.5% DF 7% 5% Crampes 2% 10% Malaise 10% Troubles du rythme 5% Chondrocalcinose 7.5% 7% Polyuropolydipsie 20% Déshydratation 10% Vomissements 13% Constipation 7% p<0.0001 p=0.037 n=85, Med. IQR 30 (28-32) n=25, Med. IQR 32.9 (28.5-36) n=61, Med. IQR 97 (94.5-99) n=25, Med. IQR 89 (80-96.5) p<0.0001 p<0.0001 n=89, Med. IQR 0.59 (0.51-0.68) n=26, Med. IQR 0.87 (0.77-0.96) n=81, Med. IQR 0.05 (0.03-0.11) n=21, Med. IQR 0.62 (0.23-1.14) Bartter anténatal Syndrome de Bartter anténatal - Type I Na+ 2Cl K+ Furosemide K+ Na+ K+ Cl Barttine Cl K+ K+ H+ (+) Réabsorption: Na+ 20% Ca2+ 20 % Mg2+ 60-70% Ca++ Mg++ NH4+ Na+ RSCa Na+ Claudines 16 et 19 HCO3- Vte (-) Gène SLC12A1 – 15q15-21 (NKCC2) 18% ** 54% 10% * 5% 13% Na-K 2-Cl : 1.099 aa. Gène: 26 exons N = 30 : 16 homozygotes, 10 hétérozygotes comp., 4 une mutation hétérozygote Missense Splicing Nonsense Frameshift Inframe Syndrome de Bartter anténatal - Type II Na+ 2Cl K+ Furosemide K+ Na+ K+ Cl Barttine Cl K+ K+ H+ (+) Réabsorption: Na+ 20% Ca2+ 20 % Mg2+ 60-70% Ca++ Mg++ NH4+ Na+ RSCa Na+ Claudines 16 et 19 HCO3- (-) Hyperkaliémie transitoire Gène KCNJ1 – 11q24-25 (ROMK) Evolution Ionogramme – Bartter de type II 160 140 120 Na Cl 100 80 60 0 2 4 6 8 10 12 14 16 18 20 22 24 30 27,5 25 22,5 20 17,5 K RA 15 12,5 10 7,5 5 2,5 0 0 2 4 6 8 10 12 14 Jours 16 18 20 22 24 21% Faux sens Non sens Frameshift 7% 72% Canal K Kir 1.1. : 391 aa. Gène: 5 exons N = 34 : 15 homozygotes, 17 hétérozygotes composites, 2 une seule mutation htz. Syndrome de Bartter anténatal avec surdité Na+ 2Cl K+ Furosemide K+ Na+ K+ Cl Barttine Cl K+ K+ H+ (+) Réabsorption: Na+ 20% Ca2+ 20 % Mg2+ 60-70% Ca++ Mg++ NH4+ Na+ RSCa HCO3- Na+ Claudines 16 et 19 (-) Type IV - Gène BSND – 1p31 Syndrome de Bartter anténatale avec surdité - Type IV BSND – 1p31 N =12 9 homozygotes 3 hétérozygotes composites. CLCNKA-CLCNKB – 1p36 Hérédité Digénique Bartter IVb Barttin. : 320 aa. Bartter IVa Rickheit G et al EMBO 2008 Syndrome de Bartter anténatal Brochard, K. et al. Nephrol. Dial. Transplant. 2009 24:1455-64 Syndrome de Bartter anténatal KCNJ1 SLC12A1 CLCNKB BSND Nombre 23 17 6 6 Médiane 25 26 28 22 KCNJ1 SLC12A1 CLCNKB BSND Nombre 31 28 16 7 Médiane 32 32 37 31,8 * p = 0,021 ** p = 0,002 *** p = 0,001 Syndrome de Bartter anténatal KCNJ1 SLC12A1 CLCNKB BSND Nombre 32 18 10 7 Médiane 1321,5 131 131 121 KCNJ1 SLC12A1 CLCNKB BSND Nombre 28 21 14 9 Médiane 5 3 2.6 2.4 * p = 0,002 ** p = 0,0001 *** p = 0,0006 Syndrome de Bartter anténatal KCNJ1 SLC12A1 CLCNKB BSND Nombre 18 15 9 7 Médiane 26.5 28 39 27 * p = 0,01 ** p = 0,002***p=0,003 KCNJ1 SLC12A1 CLCNKB BSND Nombre 17 10 7 6 Médiane 92 89 76 83 * p = 0,01 Syndrome de Bartter anténatal Conclusion Main biological and clinical characteristics and genetic classification Parameter Antenatal Bartt e r Classic Bartte r Type I Type II Type IV Age of presentation In utero In utero In utero In utero Polyhydramnios/ Prematurity Failure to thrive Deafness Polyur i a Tetany crisis Magnesemia +++ +++ +++ +++ +++ Normal +++ +++ Normal - Transient Hyperkalemia Hypochloremic alkalosi s Calciur i a Nephrocalcinosis Urinary prostaglandins Indomethacin response Gitelman Type III Adolescent/adult /children ++ Infant- toddler, preschool or school children - +++ Yes +++ Normal +++ +++ Normal ++ ++ Occasional Normal or low +++ - - - + Commo n Low (sometimes normal) - ++ ++ +++ High +++ High +++ +++ High +++ +++ High +++ Normal or High + ++ High +++ +++ +++ +++ - ++ ++++ Normal or High + Low or normal High +++ +/Normal or High + Normal +++ ++ + EAST : Epilepsy, Ataxia, Sensorineural Deafness, Tubulopathy Bockenhauer D et al. New Engl J Med 360, 2009 SeSAME: Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance Scholl UI et al. PNAS 106, 2009 Tubule distal Cl Na+ Thiazides TRPV5 TRPM6 Na+ K+ Ca2+ K+ Mg2+ Ca2+ K+ H+ Réabsorption: Na+ 7% Ca2+ 10- 15% Mg2+ 10% Cl- Na+ Na+ Ca2+ Na+ Mg2+ Syndrome EAST ou SeSAME Gène KCNJ10, 1q23.2 Bartter type I - Furosémide 2Cl Na+ K+ Furosémide K+ (+) HCO3- Cl K+ Na+ Na+ Ca++ Mg++ NH4+ HCO3- Barttine K+ H+ Pendrine Na+ K+ Cl - Cl- HCO3- Na + Cl- K+ ClCl- H+ HCO3- Intercalaire B Na + NDCBE Claudines (-) HCO3 H+ H+ Vte HCO3Cl- Intercalaire A ACII Bartter type IV –Furosémide/Thiazide H+ K+ Na + K+ CO2 Bartter type II – Furosémide/Amiloride H2O (-) (+) ENaC Bartter type III – Thiazide/Furosémide Amiloride Na + Na + K+ Principale Aldostérone K+ K+ Na+ Ca2+ Na+ Mg2+ H+ Seyberth HW, Ped Nephrol 2011 Cl- Cl Na+ Thiazides Na+ Na+ Na+ Ca2+ H2O RM Ca2+ Mg2+ Gitelman/East - Thiazide Spironolactone Eplérenone H2O Récepteur sensible au calcium (RSCa) BLAH BLAH > AMPc > > + Na+ 2ClK+ - RSCa 3q21-24 + Ca2+ (Mg2+) - K+ Ca2+ Mg2+ ADH, PTH + _ Cl- Hypocalcémie Autosomique Dom. •PTH basse, inappropriée •Hypomagnésemie •Hyperphosphatémie •Lithiase/néphrocalcinose •Polyurie Association HAD et Bartter • • • • • Réponse normalisée Polyurie Hypomagnésémie Hypercalciurie Alcalose hypokaliémique Hyperéninémie et hyperaldostéronisme modérées • Hypoparathyroïdie • EFR 1.0 0.9 0.8 0.7 0.6 0.5 0.4 0.3 0.2 0.1 0.0 0 2 4 6 8 10 12 14 16 Ca2+ (mM) ERK activity (% of WT activity) 150 RFD de Cl: 71 % (N=86.7+4.1 %) Osmol. Ur. Min. 90 mOsm/kg (N=54+13) Osmol. Ur. Max. 460 mOsm/kg (N=>800) Receptor WT L125P I40F 1.1 WT L125P I40F 100 EC50 3.2 mM 1.4 mM 8.1 mM Hill Slope 4.1 2.0 3.1 Bartter V 50 0 -3.5 -3.0 -2.5 -2.0 -1.5 Log [Ca +2 ] M Vargas-Poussou R et al. JASN 2006 Diagnostic différentiel – Dossier 1 • Dossier et prélèvement adressés pour diagnostique génétique d’un pseudohypoaldostéronisme de type 1 • Fille née le 22/03/2008, prématurée - 35 SA. Grossesse compliqué par hydramnios. PN:1.92kg TN: 44.5 cm • A J6 Tableau de déshydratation avec hyponatrémie, hyperkaliémie et polyurie • Prélèvements pour bilan hormonal et début d’un traitement par fludrocortisone 2 Tubulopathies avec perte de sel Pseudohypoaldostéronis me de type 1 Bartter type2 Hydramnios Rare Constant Fuite sodée Oui Oui Polyurie Oui Oui Persistante Transitoire Très élevées Très élevées Hyperkaliémie Rénine et aldostérone • • • Clinique en faveur d’un syndrome de Bartter de type 2 Analyse du gène KCNJ1 Détection d’une mutation connue à l’état homozygote une semaine après la réception du prélèvement • Adaptation du traitement Diagnostic Différentiel - SB anténatal Dossier 2 Grossesse • Parents d’origine tunisienne, cousins germains • 34 SA : Hydramnios + distension diffuse d’anses digestives • Naissance à 36 SA • LA teinté. Méconium émis à la naissance • A 5 mois hospitalisation et bilan pour stagnation pondérale – Poids 5250 g; Taille 61 cm • Bilan: Sang Nouveau-né • Apgar 10/10 • PN: 2970 g, TN: 49 cm • Ballonnement abdominal • • Urines Na 130 mmol/l <20 mmol/l K 2.54 mmol/l 53 mmol/l Cl 80 mmol/l RA 38 mmol/l Ca 2.6 mmol/l Ca/Cret 0.35 Mg 0.89 mmol/l Créatinine 20 µmol/l Rénine active 1114 pg/ml Aldostérone 888 pg/ml Ecographie Rénale: normale Pas de polyurie Sous NaCl 12 mEq/j et KCl 24 mEq/j Bilan: Sang Urines Na 135 mmol/l 24 mmol/l K 3.5 mmol/l 28 mmol/l Analyse des haplotypes - Bartter anténatal – Dossier 2 Diagnostic Différentiel - SB anténatal – Dossier 2 • A 1 an - diarrhée sévère à rotavirus. Bilan sous NaCl 20 mEq/kg/j et KCl 18 mEq/kg/j • P 7615 g (-1.75 DS); T 71.5 cm (-0.5 DS) • Bilan: Sang Na 144 mmol/l K 3.3 mmol/l Cl 96 mmol/l RA 33 mmol/l • A distance de l’épisode: ionogramme de selles Urine 217 mmol/l 68 mmol/l 184 Diarrhée Chlorée congénitale : Maladie autosomique récessive SLC26A3 - 7q31.1 Cl dans les selles > 90 mmol/l Na 82 mmol/l (30-50) K 28 mmol/l (50-80) Cl 131 mmol/l (30-50) Wedenoja S et al. Aliment Pharmacol Ther. 2010 And Human Mut 2011 Diagnostic Différentiel - SB classique/SG – Dossier 3 • • • • Parents non consanguins Né à terme, pas d’antécédents Mange peu et boit beaucoup Episodes de déshydratation à 8 et 10 mois • Bilan lors de ces épisodes • Sang Urines Na 132 et 128 mmol/l inadapté K 2.4 et 2.7 mmol/l 89 mmol/l Cl 79 mmol/l Ca 2.56 0.15mmol/mmol RA 42 et 40 mmol/l Rénine active 316 pg/ml Aldostérone 1120 pg/ml Ecographie Rénale: normale • A 1 an sous NaCl 1,5 g/j et K 45 mg/j P 9.5 kg (-1 DS); T 74 cm • Bilan: Sang Urines Na 139 mmol/l 29 mmol/l K 4.8 mmol/l 40 mmol/l Cl 110 mmol/l RA 23 mmol/l Ca 2.57 mmol/l Ca/Cret 0.15 Mg 0.94 mmol/l Créatinine 55 µmol/l Rénine et Aldo normales Diagnostic Différentiel - SB classique/SG – Dossier 3 • • Analyse du gène CLNKB: absence de mutation Analyse du gène SLC12A3: une seule mutation à l’état hétérozygote. Absence de grand réarrangement • En 2011: retard de croissance – Test de la sueur positive – Confirmation d’une mucoviscidose • Mucoviscidose • Maladie autosomique récessive • 1/3000 • CFTR - 7q31.2 • Pseudo-Bartter • pertes chroniques de NaCl dans la sueur • pertes extra rénales additionnelles – maladies intercurrentes – conditions environnementales particulières (canicule) Diagnostic Différentiel - SB classique/SG – Dossier 4 • • • • • • • • Garçon de 14 ans Parents non consanguins Né à terme, pas d’antécédents Douleurs musculaires (1 an) Fatigabilité Episode de douleurs musculaires sévères après sport et faiblesse musculaire sévère avec chutes le 21/09/2010 58 kg TA 120/64 Sang Urines Na 141 mmol/l 28 mmol/l K 2.1 mmol/l 29 mmol/l Cl 79 mmol/l RA 28 mmol/l Ca 2.29 Mg 0.82 mmol/l CPK 404 UI/l Rénine active 7.38 pg/ml Aldostérone 159 pmol/l sous perfusion Ecographie Rénale: normale 26/09/2010 25/01/2011 Sang Urine Sang (mmol/l) (mmol/j) Na 139 55 Cl 103 90 K 3.6 134 RA 25 Mg 0.73 5.38 0.77 Ca 2.43 2.5 2.50 FE 0.2% 3.5 9% 27 Ca/cr0.18 Créat 64 µmol/l 14 C 22.6 D 32.6 Renine (pg/ml) C 124 D 1531 Aldo (pmol/l) Sans traitement Kaléorid 1cp/j Paralysie Périodique Hypokaliémique • Prévalence 1/100.000 • Autosomique dominante – pénétrance incomplète • Seconde décennie • Manifestations après l’exercice ou un repas riche en glucides ou en Na • 70% des cas, mutations du gène du canal calcium musculaire CACNL1A3 – 1q32.1 • 10% des cas, mutations du gène du canal sodium musculaire SCN4A – 17q23.3 Fontaine B., Adv Genet, 2008 BAN115001 • Premier enfant, famille Greque non consanguine • Fille , 2 ans • Retard de croissance • Grossesse : hydramnios 33 SA • Née à 37 SA • Poids : 2550 gr • Pas de manifestations cliniques en période néonatale • Néphrocalcinose Na 139 mmol/l K 3.2 mmol/l Cl 91 mmol/l HCO3 32.1 mmol/l Protéines 82 g/l Ca 2.8 mmol/l Mg 1.19 mmol/l créat 25 µmol/l Rénine 523 pg/ml Aldo 30 ng/dl PTH 76.9 pg/ml 25OHvitD 23.2 ng/ml Urines K 43 mmol/l Cl 14 mmol/l Ca/Créat 0,41 mmol/mmol BAN 115 CLCNKA-CLCNKB – 1p36 ATG ATG Absence d’amplification par PCR des exons 2 - 6 Délétion homozygote E2_E6 Exons 7 à 20 Séquences normales 0 CLCNKA CLCNKB Mapview Sondes control c c c c c c c c c c c c c c c 01-016.2 CLCNKB Exon 19 01-016.2 CLCNKB Exon 18 01-016.2 CLCNKB Exon 17 01-016.2 CLCNKB Exon 15 01-016.2 CLCNKB Exon 14 01-016.2 CLCNKB Exon 13 01-016.2 CLCNKB Exon 11 01-016.2 CLCNKB Exon 10 01-016.2 CLCNKB Exon 08 01-016.2 CLCNKB Exon 06 01-016.2 CLCNKB Exon 05 01-016.2 CLCNKB Exon 03 01-016.2 CLCNKB Exon 02 01-016.2 CLCNKB Exon 01 01-016.2 CLCNKA Exon 10 01-016.2 CLCNKA Exon 05 01-015.7 CASP9 01-014.0 PRDM2 Ratio BAN 115 - patient D02_ban115001_caro020312.fsa^GMsTXT.txt 1,2 1 0,8 0,6 0,4 0,2 0 CLCNKA CLCNKB Mapview Sondes control c c c c c c c c c c c c c c c 01-016.2 CLCNKB Exon 19 01-016.2 CLCNKB Exon 18 01-016.2 CLCNKB Exon 17 01-016.2 CLCNKB Exon 15 01-016.2 CLCNKB Exon 14 01-016.2 CLCNKB Exon 13 01-016.2 CLCNKB Exon 11 01-016.2 CLCNKB Exon 10 01-016.2 CLCNKB Exon 08 01-016.2 CLCNKB Exon 06 01-016.2 CLCNKB Exon 05 01-016.2 CLCNKB Exon 03 01-016.2 CLCNKB Exon 02 01-016.2 CLCNKB Exon 01 01-016.2 CLCNKA Exon 10 01-016.2 CLCNKA Exon 05 01-015.7 CASP9 01-014.0 PRDM2 Ratio BAN 115 - père E02_ban115010_caro020312.fsa^GMsTXT.txt 1,4 1,2 1 0,8 0,6 0,4 0,2 0 CLCNKA CLCNKB Mapview Sondes control c c c c c c c c c c c c c c c 01-016.2 CLCNKB Exon 19 01-016.2 CLCNKB Exon 18 01-016.2 CLCNKB Exon 17 01-016.2 CLCNKB Exon 15 01-016.2 CLCNKB Exon 14 01-016.2 CLCNKB Exon 13 01-016.2 CLCNKB Exon 11 01-016.2 CLCNKB Exon 10 01-016.2 CLCNKB Exon 08 01-016.2 CLCNKB Exon 06 01-016.2 CLCNKB Exon 05 01-016.2 CLCNKB Exon 03 01-016.2 CLCNKB Exon 02 01-016.2 CLCNKB Exon 01 01-016.2 CLCNKA Exon 10 01-016.2 CLCNKA Exon 05 01-015.7 CASP9 01-014.0 PRDM2 Ratio BAN 115 - mère F02_ban115011_caro020312.fsa^GMsTXT.txt 1,2 1 0,8 0,6 0,4 0,2 Hypokaliémie d’origine rénale I 1 II 2 1 3 2 4 3 Famille française non-consanguine avec 5 individus atteints sur deux générations. Phénotype Plasma Na+ K+ Unités Cl - HCO3 - Urines Mg mmol/l Rénine* Aldostérone* mU/l pmol/l Na+ K+ Cl- Mg Ca mmol/24h Ca/Créatinine mmol/mmol Normes 135-145 3,5-4,5 95-105 22-27 0,71-1,04 15-50 208-100 II-2 Cas index 140 2,6 94 33 0,52 272 943 258 119 286 2,9 2,5 0,16 II-1 Sœur atteinte 138 2,9 99 29 0,62 197 1961 123 87 106 0.7 0.12 0,01 II-3 Sœur 137 3,5 101 26 0,9 I-1 oncle atteint 138 2,5 92 37 0,49 133 1488 245 179 345 6,2 5,8 0,51 I-2 père 140 4.3 103 29 0.87 I-3 Mère 137 2,2 95 33 0,58 124 596 166 128 243 3,8 3,85 0,41 I-4 tante atteinte 136 2,2 94 36 0,64 104 917 247 27 324 2,1 3,38 0,4 * Après une heure d’orthostatisme Séquençage du gène SLC12A3 I Mutation 1 1 2 3 Mutation 2 Mutations 3 et 4 Mutations 2,3 et 4 II 4 1 Mutations 1,3 et 4 2 Hétérozygote comp. Mutation 1 et 2 3 Mutations 3 et 4 Mutation 1, exon 10: c.120insGTGATGC, p.Asp400X Mutation 2, exon 23: c.2747+1G>A Mutation 3, exon 14: c.1805_1806del, p.Tyr602CysfsX31 Mutation 4, exon 22: c.2660+1G>A Mutations 2,3 et 4 Séquençage du gène SLC12A3 1 I 3 4 2 3 1 2 3 4 1 II 1 3 4 2 1 2 4 3 4 2 3 3 4 Mutation 1, exon 10: c.120insGTGATGC, p.Asp400X Mutation 2, exon 23: c.2747+1G>A Mutation 3, exon 14: c.1805_1806del, p.Tyr602CysfsX31 Mutation 4, exon 22: c.2660+1G>A 2 Traitement Na+ Cl Na+ Thiazides 7% Gitelman ClK+ Na+ Na+ Ca2+ Ca2+ Mg2+ Na+ H+ Na+ Ca2+ Na+ Mg2+ Suppléments en sel, K, Mg Diurétiques (amiloride) Na+ 20% 2Cl Na+ K+ Furosémide K+ K+ Ca++ Mg++ NH4+ (+) H+ Na+ Na+ K+ Cl Barttin Cl K+ HCO3- Na+ (-) Vte Bartter Prise en charge périnatale Suppléments en sel, K, Mg Indométacine PHRC- GITAB- Dr Anne BLANCHARD - Hommes ou femmes âgés de 18-60 ans, - atteints de syndrome de Gitelman génétiquement confirmé. - Contraception efficace chez les femmes en âge de procréer. S’il s’agit d’une contraception œstro-progestative maintenue inchangée pendant toute l’étude. - Consentement éclairé signé Mesure de kaliémie, natrémie, créatininémie en ville MODAMIDE 20 10 5 CHRONOINDOCID (+ oméprazole 20 mg) INSPRA 150 100 50 75 REPONSE AUX TRAITEMENTS 100 Intolerant Non responder Intermediate responder Full responder Patients (%) 80 60 40 20 0 Indomethacin (n=30) Eplerenone (n=30) Amiloride (n=30) Delta kaliémie < 0,1 mmol/l : non répondeur 0,1-0,3 mmol/l : répondeur intermédiaire > 0,3 mmol/l : répondeur CONCLUSION - EFFICACITE : Indometacine> eplerenone= amiloride - TOLERANCE COURT TERME: eplerenone> amiloride>Indometacine - A LONG TERME: Risques avec indometacine : rénal, digestif, cardiovasculaire Eplerenone: prise de poids, troubles sexuels Amiloride à cette dose: troubles du sommeil, douleurs abdominales INDICATION A POSER EN FONCTION DU RAPPORT BENEFICE/RISQUE Signification clinique de l’hétérozygotie pour des mutations du gène SLC12A3 PHRC 2013, HEPHYGI Pression artériellle : Population : Patient Gitelman -/Pat. hétérozygotes +/Contrôles +/+ Auto mesure, ECG n= 50 n=100 n=100 National HEGP : CRC, Pr M Azizi, Dr A Blanchard, Pr P. Houillier Hôpital Tenon: Pr JP Haymann CHU Limoges: Pr M. Essig HC Lyon: Dr L. Doubourg CHU Toulouse: Pr I. Tack Homéostase Na K : Sang, Ur 24h, rénine, aldostérone Homéostase Mg, Ca : Sang, Ur 24h Urine, marqueurs osseux Vaisseaux : Mesure de la PAC et de la VOP Marqueurs ADMA/SDMA NO2/NO3 Ur Metabolisme : Poids, taille, BMI HGPO; IRI Métabolisme de lipide Sérothèque et Urothèque Signification clinique de l’hétérozygotie pour des mutations du gène SLC12A3 PHRC 2013, HEPHYGI H de J Utilisation appareil d’automesure Organisation dates Consentement BP ECG Prise de sang HGPO SphygmoCor Mesure PA Recueil Ur 24 h J1 V0 Information J2 J3 V1 MERCI Centres de Référence Centres de Compétences La Réunion
© Copyright 2025 Paperzz