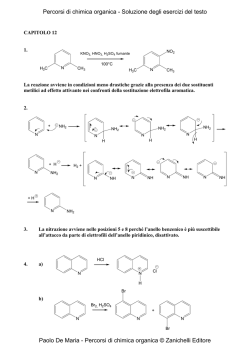
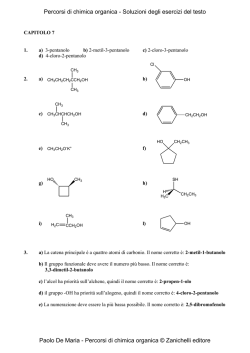
The Sicilian e-Infrastructure: COMETA Consortium Simulation and Theory of Polar/Apolar Fluid Mixtures D. Costa1, F. Saija2, G. Munaò2, C. Caccamo2 1 Consorzio COMETA and Dip. di Fisica, Università di Messina, Ctr. Papardo – 98166 Messina (dino.costa@unime.it) 2 CNR- Istituto Processi Chimico-Fisici, Contrada. Papardo, Salita Sperone – 98158 Messina 3 Dipartimento di Fisica, Università di Messina, Contrada Papardo – 98166 Messina Molecular Dynamics simulation and Reference Interaction Site Model (RISM) theory have been used to study fluid mixtures composed by methanol (CH3OH) and carbon tetrachloride (CCl4), focusing in particular on the low concentration regime of methanol. Simulations clearly show the modifications in the associating properties of the methanol induced by the presence of the apolar solvent: the liquid structure of pure methanol is characterized by winding chains of hydrogen-bonded monomers; upon dilution, the methanol tends to preserve its local order, whereas a significant fraction of chains closes to form cyclic structures, with important implications for the thermodynamic properties of the system. Theoretical results qualitatively follow the simulation trends. An analysis of performances on the GRID architecture of the Consorzio COMETA is shortly presented. ' $ ' Motivations ' $ Molecular Geometries Interaction Potentials The interaction potential between any site α on a molecule and another site β on a different molecule at distance r apart is written as: »“ – qα qβ σαβ ”12 “ σαβ ”6 LJ coul + vαβ (r) ≡ vαβ (r) + vαβ (r) = 4ǫαβ − r r r Monohydric alcohols and their mixtures with nonpolar solvents constitute a class of complex fluids particularly useful for the investigation of the selfaggregation properties of liquids interacting via hydrogen bonds [1] The CH3 OH/CCl4 mixture in particular has been characterized by means of several experimental [2,3] and theoretical tools (see e.g. [4] and references). The purpose of the present study is manyfold: Methanol parameters – the simulation of relatively large systems, accessible through the GRID facilities, allows us to address the crucial issue of the modification of the aggregation properties of methanol in the mixture, at the microscopic scale and even in the high dilution regime. – A comparison is planned between our simulations and the experimental data generated in the IPCF laboratory of CNR [5]. Simulations will also provide the necessary benchmark to gauge the accuracy of the liquid state theories for molecular fluids currently adopted by the group at the Physics Department [6]. & Geometry of the CCl4 (left) and CH3 OH (right) models. The C site of the methanol represents the CH3 group. All diameters roughly correspond to the “collisional diameters” σXX reported in next table. The distances among the interaction sites are: LCH3 O = 1.4246 Å, LOH = 0.9451 Å, and LCH3 H = 1.9437 Å (∠ CH3 OH = 108.5◦ ) for CH3 OH; LCCl = 1.766 Å (∠ ClCCl = 109.5◦ ) for CCl4 . $ ' Plan of Simulations qCH3 qO -0.265 0.700 qH ǫCH3 CH3 ǫOO σCH3 CH3 σOO -0.435 0.2070 0.1700 3.7750 3.0710 Carbon Tetrachloride parameters % & 1 ' $ 2 q ǫCC ǫClCl σCC σClCl 0 0.1016 0.2034 4.600 3.500 Partial charges q are in e units, the potential well depth ǫ is in K cal/mol and the “collisional diameters” σ in Å. Data for CH3 OH and CCl4 are taken from [7] and [8], respectively. Lorentz-Berthelot rules apply for all cross-interactions. & % 3 ' $ Results II - Microscopic configurations Results I - Local order in the fluid % $ Parallel Molecular Dynamics package MOLDY [9] with: Molecules handled as rigid bodies using the quaternion algorithm 40 5 40 5 χM=0.1 χM=0.1 NVT simulations at room conditions via the Nosè-Hoover thermostat Ewald summation for the long-range part of interaction 4 χM=0.5 4 χM=0.5 30 30 χM=0.9 χM=0.9 3 g(r) Link cell with no minimum image convention 20 NCH3 OH NCCl4 Ntot MD steps 0.1 876 7872 41988 40 000 0.5 1458 1458 11664 300 000 0.9 2624 292 9332 40 000 0 4 ' Ideal Scaling 10 True Scaling True Scaling Log(Time) Time (mins) 1000 8 500 6 0 0 20 40 # CPU’s 60 4 0 2 4 6 Log(# CPU’s) Running time vs the number of CPU. The structure of the problem at issue (where the sample size is large in comparison with the typical spatial range of molecular interactions) produces an almost ideal scaling of the computer time. In this case in fact, a proper partition scheme of the simulation box into several subsystems minimizes the communication requirements among the parts of the sample, and this process in turn optimally copes with a parallel split of the computational load among the various processors. & 3 5 7 10 OO 1 0 0 2 4 6 1 8 0 r[A] Radial distribution functions (RDF) of O and H sites of methanol (in black) and corresponding coordination numbers (CN, in red), at several methanol concentrations. The sharp increase of RDF accompanied by the constancy of CN reflect the tendency of the methanol to preserve the local environment typical of the pure liquid, notwithstanding the progressive dilution of such species [4]. % & 5 Conclusions and perspectives Extensive MD simulations of liquid mixtures formed by CH3 OH and CCl4 at different concentrations are presented. The use of computer simulations provides a powerful tool to understand the origin of the self-association phenomena driven by mutual hydrogen-bond interactions, from a detailed microscopic point of view. 12 Ideal Scaling 1 $ ' Analysis of performances 1500 OH r[A] χM is the concentration of methanol. NCCl4 and NCH3 OH are the number of molecules of carbon tetrachloride and methanol, respectively. Ntot is the total number of interaction sites. & 2 2 10 χM N(r) 3 20 The computing facilities available at the Consorzio COMETA open the possibility to deal with larger systems and longer time trajectories, in comparison with the typical sizes investigated in the past [4, 7, 8]. This opportunity allows the investigation of physical properties otherwise practically unaccessible with normal computational resources. – The accurate determination of the microscopic behaviour of the methanol in the low concentration regime is one of these aspects. – Another important issue concerns the long-range behaviour of the correlation functions. This is related to the structure factors, directly calculated in experiments, and to the macroscopic properties of the system through the compressibility route to thermodynamics. % & Snapshots of equilibrated configurations. For clarity sake, only the O (in red) and H (in white) sites of methanol are drawn. In the equimolar mixture (left), the formation of linear chains provides the dominant mechanism of aggregation of methanol molecules. At low concentration (χM = 0.1, right) chains tend to form cyclic structures (rings), with an evident increase of isolated monomers. & % ' $ 6 References % $ [1] C. A. Angell in Hydrogen Bonded Liquids, J. C. Dore and J. Texeira eds. (Kluwer Academic Publishers, Dordrecht, 1991). [2] G. C. Paraskevopoulos and R. W. Missen, Trans. Faraday Soc. 57, 869 (1962); J.-E. A. Otterstedt and R. W. Missen, Trans. Faraday Soc. 57, 879 (1962). [3] M. Musso, H. Torii, P. Ottaviani, A. Asenbaum, and M. G. Giorgini, J. Phys. Chem. A 106, 10152 (2002). [4] R. Veldhuizen and S. W. de Leeuw, J. Chem. Phys. 105, 2828 (1996). [5] M. Pieruccini, F. Saija, C. Vasi, and M. Musso, Chem. Phys. Lett. 382, 523 (2003); F. Aliotta, M. Musso, R. Ponterio, F. Saija, and G. Salvato, J. Phys. Chem. B 108, 12972 (2004); B. Fazio, M. Pieruccini, and C. Vasi, J. Phys. Chem. B 109, 16075 (2005). [6] D. Costa, G. Munaò, F. Saija, and C. Caccamo, J. Chem. Phys. 127, 224501 (2007). [7] W. L. Jorgensen, J. Phys. Chem. 90, 1276 (1986). [8] I. R. McDonald, D. G. Bounds, and M. L. Klein, Mol. Phys. 45, 521 (1982). [9] K. Refson, Comp. Phys. Comm. 126, 309 (200); see also the webpage www.earth.ox.ac.uk/∼keithr/moldy.html. % & 7 8 9 Acknowledgment: This work makes use of results produced by the PI2S2 Project managed by the consorzio COMETA, a project co-funded by the Italian Ministry of University and Research (MIUR) within the Piano Operativo Nazionale Ricerca Scientifica, Sviluppo Tecnologico, Alta Formazione (PON 2000-2006). More information is available at http://www.pi2s2.it and http://www.consorzio-cometa.it %
© Copyright 2025 Paperzz